CARMONA Lab
Our focus
We study patterns of variation across cancer patients to identify general principles of immune system regulation during tumor progression and response to therapy. We combine innovative data science with high-throughput single-cell and spatial omics technologies with the goal to reveal biological insight and develop predictive models of disease progression and response to treatment.
Our projects
Advancing single-cell omics data analysis
Recent advances in single-cell and spatial omics technologies have opened a unique opportunity to explore biological systems at an unprecedented resolution and scale. Our lab develops computational and statistical methods to translate the complex data generated by these technologies into biological understanding. We have created many widely-used open-source computational methods tackling multiple challenges in single-cell data science including integration, cell type classification, reference atlas mapping, gene signature scoring, gene program discovery, and T cell clonal analyses.

Dissecting the tumor microenvironment variation across and within individuals
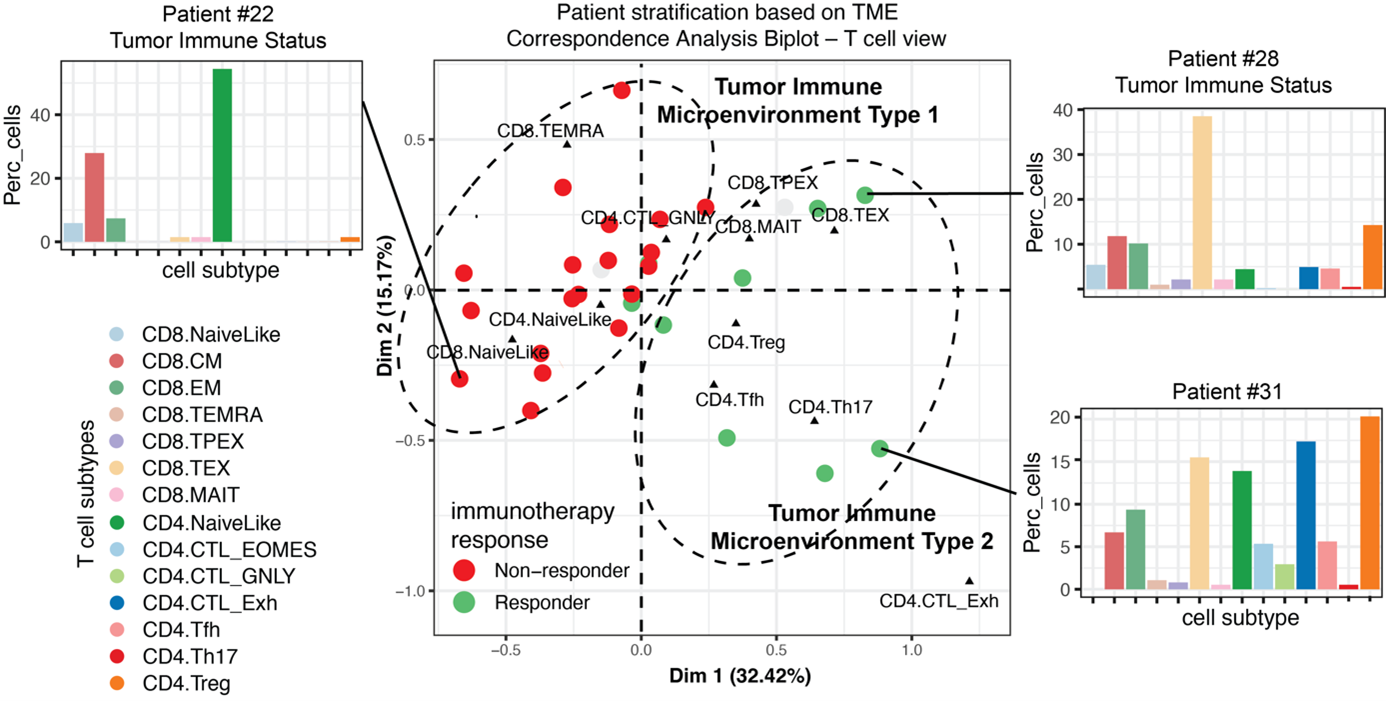
The tumor microenvironment (TME) is the complex niche surrounding cancer cells, composed of various cell types and molecules that influence tumor growth and treatment responses. In the TME, a striking diversity of immune cells, fibroblasts, endothelial cells, and other tissue-specific cells interact with phenotypically diverse cancer cells and evolve with cancer progression. The TME components and their complex interaction network determine to a large extent disease progression and response to therapies. We use high-resolution spatial and single-cell technologies to study the biological variation that occurs within and between tumors, with the goal to identify general principles of immune system regulation. This approach has the potential to reveal the predominant immune regulation mechanisms underpinning cancer progression and therapy resistance, leading to novel therapeutic targets.

Predictive models for immuno-oncology
Therapies that modulate the immune system, such as immune checkpoint inhibitors, have revolutionized the landscape of cancer treatment. However, only a fraction of patients experience clinical benefit across cancer. Profiling the tumor immune microenvironment offers a great opportunity for the discovery of biomarkers of therapy response and of immune-related adverse events. However, tumor biopsy-based biomarkers face limitations due to tumor heterogeneity and accessibility of various metastatic sites. In contrast, peripheral blood samples present several advantages including enhanced accessibility, feasibility to implementation in clinical practice and possibility of multiple sampling for monitoring of dynamic changes.
We study how the peripheral blood is altered by the tumor characteristics at the single-cell level and exploit this knowledge to develop machine learning models to predict disease state and response to therapy.
Defining transcriptional and epigenetic reference maps of immune cells
Single-cell transcriptomics and chromatin accessibility profiling allow us to unbiasedly explore cell diversity and to discover cell types, transient cell states and their characteristic gene programs. Robust, expert-curated single-cell maps that can be created with these data modalities have become essential resources as they serve as references to interpret cellular differences across perturbations, conditions, tissues and individuals. A single-cell map is a useful abstraction that allows modeling complex cell heterogeneity as discrete or partially-overlapping cell populations defined by characteristic gene programs activity and chromatin accessibility patterns – and potentially distinct functions. Combining our expertise in bioinformatics and immunology, we generate high-quality reference maps for immune cells, which are highly used by the biomedical research community. These are available at the SPICA portal https://spica.unil.ch/.

KEY PUBLICATIONS

-
Massimo Andreatta, Leonard Herault, Paul Gueguen, David Gfeller, Ariel J Berenstein, Santiago J Carmona. Semi-supervised integration of single-cell transcriptomics data. Nature Communications (2024)
-
Corria-Osorio J, Carmona SJ, Stefanidis E, Andreatta M, Seijo B, Scarpellino L, Ronet C, Muller T, Luther S, Irving M, Coukos G. Orthogonal cytokine engineering enables novel synthetic effector states escaping canonical exhaustion in tumor-rejecting CD8+ T cells. Nature Immunology (2023)
-
Massimo Andreatta, Zachary Sherman, Ariel Tjitropranoto, Michael C. Kelly, Thomas Ciucci, Santiago J. Carmona. A single-cell reference map delineates CD4+ T cell subtype-specific adaptation during acute and chronic viral infections. eLife (2022)
-
Andreatta M, Gueguen P, Borcherding N, Carmona, SJ. T Cell Clonal Analysis using Single-Cell RNA Sequencing and Reference Maps. Bio-protocol journal (2023)
-
Massimo Andreatta, Ariel J Berenstein, Santiago J Carmona. scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets. Bioinformatics (2022)
- Massimo Andreatta, Fabrice P.A. David, Christian Iseli, Nicolas Guex, Santiago J.Carmona SPICA: Swiss Portal for Immune Cell Analysis. Nucleic Acids Research (2022)
- Andreatta M and Carmona SJ. UCell: Robust and scalable single-cell gene signature scoring. Computational and Structural Biotechnology Journal (2021)
-
M Andreatta, JC Osorio, S Muller, R Cubas, G Coukos, SJ Carmona. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nature Communications (2021)
-
Andreatta M & Carmona SJ. STACAS: Sub-Type Anchoring Correction for Alignment in Seurat. Bioinformatics (2020)
-
Santiago J. Carmona, Imran Siddiqui, Mariia Bilous, Werner Held, David Gfeller. Deciphering the transcriptomic landscape of tumor-infiltrating CD8 lymphocytes in B16 melanoma tumors with single-cell RNA-Seq. OncoImmunology (2020)
Meet the Carmona Lab Members.
| Funding |
|
| Resources | |
|
Affiliations |
|
| Links |








")